解説1.C. elegansの基礎知識と研究方法
大島靖美(九州大学・理学部) E-mail yohshscb@mbox.nc.kyushu-u.ac.jp
A.一般的情報源
(1)教科書、参考書
①THE NEMATODE CAENORHABDITIS ELEGANS, ed. S. B. Wood et al., Cold Spring Harbor Laboratory, USA(1988). Methods, Appendices (Parts List, Neuroanatomy, Cell Lineages)が利用価値が高い。
②C. ELEGANS II, ed. D. L. Riddle et al., Cold Spring Harbor Laboratory Press, USA (1997). 最新のBibliography(文献リスト)、及びGene (mutant) Listが出ている。
③小原雄治編「線虫」、ネオ生物学シリーズ⑤、共立出版(1997)。入門書
④Caenorhabditis elegans: Modern Biological Analysis of an Organism, Methods in Cell Biology Vol 48, ed. H. F. Epstein and D. C. Shakes, Academic Press, San Diego, USA (1995). 方法。
⑤J. G. White, E. Southgate, J. N. Thomson and S. Brenner: The Structure of the nervous system of the nematode Caenorhabditis elegans. Phil. Trans. R. Soc. Lond. B Biol. Sci. 275, 327-348 (1976). ニューロンとその回路。
⑥The Structure of Nematodes, 2nd Edition, ed. A. F. Bird and J. Bird, Academic Press, San Diego, USA (1991)
(2)その他
1)Worm Breeder's Gazette. Caenorhabditis Genetics Center (CGC, 項目Cに記載)から、年間3回(February, June, October)に出されている。C. elegansを中心とする線虫の情報誌。現在の最新号はVol 14, No. 5 (Feb, 1997)で、次回はmap issueの予定。9月にVol. 15, No. 1が出される。購読(subscription)はCGCに申し込む。Vol. 15のsubscription feeは未定(Vol. 14については$35だった?)。最近は各issueとともにsubscriber directory(住所録)が送られて来て、便利である。論文(A4で1頁)は投稿自由(無料)。毎号、Bibliographic References(新しい文献のリスト)もついている。
2)ACEDB:実習6(佐野)。
B.体の構造、発生、細胞
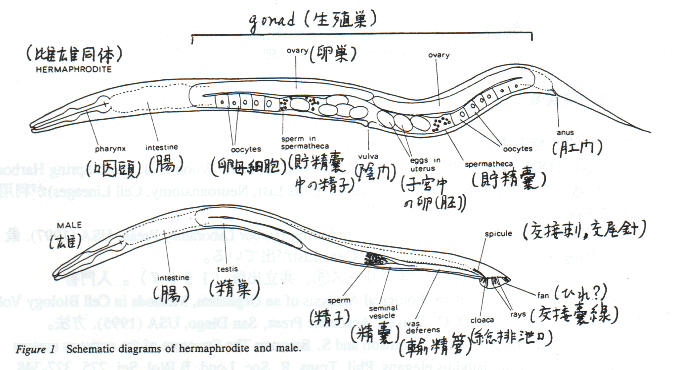
(1)体の構造と器官、組織の名称(図1、文献①、p250、J. Hodgkinより引用)。
他に、cuticle(クチクラ、外皮)、body wall muscle(体壁筋)、epidermis(上皮)、hypodermis(下皮)、nervous system(神経系)、amphid(頭部感覚子)などがある。
(2)発生
egg, embryo(胚)、hatching(孵化)、larva(幼虫、L1-L4)、molt(ing)(脱皮),
dauer larva(耐性幼虫)、adult(成虫)など。
(3)細胞と細胞系譜(cell lineage)
雌雄同体成虫のもつ体細胞(核)総数は959、生殖細胞(核)は約2,000である。
全ての体細胞には、図2のような細胞系譜に基づく系譜上の命名がなされている。例えば、陰門前駆細胞の1つP6.pはABpl/rappaappであり、anchor cell(AC)はMSpppaappppまたはMSappaapaaaである(文献①、Appendix1,3参照)。多くの細胞には上のP6.p, ACのような便宜的通称名がつけられているが、ニューロンの場合はアルファベット3文字また2文字+数字で表される(AFD, DD3など)。
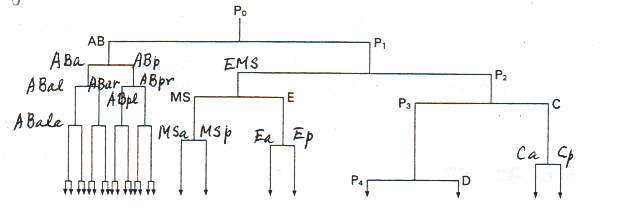
図2.細胞系譜の大略と細胞の系譜上の命名。a、pは前後分裂によって生じる前、後の細胞を、l、rは左右分裂によって生じる左、右の細胞を示す。他に、d、v(背側、腹側)の分裂もある。文献③、p25(三輪錠司)の図の一部を改変。
C.遺伝子、突然変異、変異株、表現型及び遺伝子産物の表記法及び変異株の入手
(1)遺伝子の表記
遺伝子名は、イタリックの小文字3文字、ハイフン及び数字で示す。その後ろに連鎖群(染色体)名をローマ数字(イタリック)で示す時もある。
例:dpy-1 III、unc-51 V
(2)遺伝子名の由来
unc(uncoordinated movement、非協調的運動)、dpy(dumpy、太短い)、lon(long)、sma(small)、lin(lineage)、daf(dauer larva formation)、egl(egg laying defective)、rol(roller)、vab(variable abnormal)、bli(blistered、水泡)、let(lethal)、ncl(nucleolus)など。
(3)突然変異
遺伝子名の後ろに、変異(allele)名を( )をつけて示す。allele名は小文字アルファベット1、2字(登録)+数字。
例:dpy-1(el)、unc-51(e369)V
(4)(突然)変異株(mutant、mutant strain)
単一変異株の遺伝子型は、突然変異と同じ。多重変異株の遺伝子は、突然変異を連鎖群の順に、セミコロン(;)で区切って書く。同じ連鎖群の中では染色体上の左から右へ、一字あけて書く。strain(系統、株)の名前は、大文字アルファベット2文字(登録)+数字で示す。
遺伝子型の例:flr-3(ut9)IV;flr-1(ut11)X(二重変異株)
strainの記載例:FK106(ttx-3(ks5)), CB30(sma-1(e30)), MT3353(egl-15(n484) sma-5(n678)X), DA438(bli-4I; rol-6II; daf-2 vab-7III;unc-31IV; dpy-11V; lon-2X)
ヘテロ接合体:ttx-1/unc-51(ttx-1+/+unc-51)など
(5)染色体の欠失、重複
大きな欠失をもつ染色体:mnDf62II、nDf17II、eDf2IIなどと書く。
染色体の一部が重複(duplicate)して、余分に存在するもの:dDp3III、eDp6IIIなど。
(7)遺伝子導入により生じたextrachromosomal arrayはEx10[let-23(+), ncl-1(+)]などと書く。
(8)遺伝子産物(タンパク質)
遺伝子名をローマン体の大文字で書く。
例:UNC-14、LET-23(最近まで、Unc-14のような書き方もされていた)。
(9)表現型
ローマン体で、最初の1字のみ大文字。
例:Unc、non-Dpy、Egl、Muv(Multi vulva)
(10)Strain(突然変異株)の入手
最大の入手先は下記のCGCであり、E-mail、FAX、手紙等で簡単に利用目的を書いて頼めば、2週間程度で送ってくれる(現在無料)。
Theresa Stiernagle, Caenorhabditis Genetics Center, University of Minnesota, 1445 Gortner Avenue, St. Paul, Minnesota 55108-1095 E-mail: stier@biosci.cbs. umn.edu; FAX: 1-612-625-5754 Strainのstock listもCGCから入手可能
(11)登録
遺伝子名、突然変異(allele)名、系統名はCGCに登録する。系統名または突然変異名のアルファベットが示す、それらを単離(登録)した研究室の対応表は、文献②、p887~にある。
(12)遺伝子総数など
推定遺伝子総数=14,000(文献②、p38, Waterston et al.)
突然変異により同定された遺伝子=1,400(Hodgkin et al., Science 270, 410-414, 1995)
ゲノムの大きさ=1×108塩基対、染色体=5常染色体(I~V)とX性染色体
D.ライブラリー(library)及びベクター(vector)
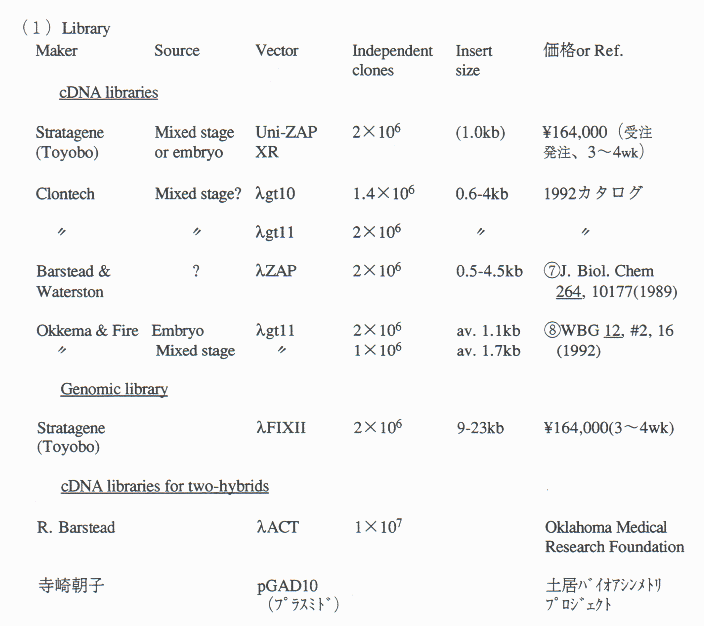
その他に、cosmid library、YAC library、YAC polytene filter(YAC grid、hybridization用)があり、cosmidまたはYACの特定のいくつかのクローン及びYAC filterは、Alan Coulson(Sanger Center, Hinxton Hall, Cambridge CB10 1SA, England, E-mail: alan@sanger.ac.uk)に目的を書いて頼めば送ってもらえる。また、小原さんもcDNA libraryを作っている。
(2)ベクター
A. Fireの研究室で、cDNA発現用のpPD21.28他多くのベクターを作成しており、頼めばもらえる。
Andrew Fire, Carnegie Institution of Washington, Department of Embryology, 115 West University Parkway, Baltimore, MD21210, USA. FAX: 1-410-243-6311, E-mail fire@mail.ciwemb.edu
文献⑨ A. Fire et al., A modular set of lacZ fusion vectors for studying gene expression in Caenorhabditis elegans. Gene 93, 189-198 (1990) 最近のFire研究室のベクターに関する情報は、L. Avery 研究室(http://eatworms.swmed.edu/(C.elegans infromation server at University of Texas Southwestern Medical Center))を経由して、ホームページの検索によって調べられる(解説8、線虫情報、佐野参照)。
1993年において、Fire研究室のベクターに用いられていたプロモーターは、hsp16(pPD49.78)、unc-54、myo-2、mec-7などであり、universal promoterとして使えるベクターは無かった。
E.遺伝子クローニング(Gene cloning)
(1)Cosmid rescue
EMSなどを用いるmutagenesisによって生じた変異に対応する野生型遺伝子をクローン化するときに用いる方法で、最も多くの例がある。まず変異を遺伝的にマップ(及びバッククロス)し、マップされた領域に対応するゲノムマップ(genome or physical map)上の領域に含まれるコスミド(数個-20個程度)を持つ大腸菌のクローンをA. Coulson(D(1)参照)から入手する。コスミドDNAを調製し、それらを単独またはいくつかまとめて変異株に導入し(実習3、微量注入による形質転換、古賀)、変異株の表現型を野生型に回復させる(rescueする)クローンを同定する。コスミドのサブクローンを用いて同様な実験を行い、当該遺伝子の存在する領域をせばめる。Genome projectによってその領域の塩基配列が決定されていれば、該当する遺伝子産物(タンパク質)が推定できる。いずれにしても、対応するcDNAのクローン化(PCR増巾、またはlibraryの選択による)と塩基配列の決定によるORF(open reading frame)の確認が必要。また、rescue活性を持つ遺伝子が、変異株の持つ変異遺伝子のサプレッサーでなく、その野生型対立遺伝子であることを示すために、変異株(できれば複数)がその遺伝子上に変異を持つことなどを示す必要がある。Rescue実験において、対象となる変異株の表現型が野生型のものと容易に区別できない場合は、形質転換のマーカー遺伝子として、rol-6d、GFP融合遺伝子(ともに優性)などを用いる(または、クローン化された適当な野生型遺伝子lin-15(+)、dpy-20(+)などをマーカーとして用い、対応するlin-15、dpy-20などとの二重変異株を導入対象株とする)。Rescue実験が成功するためには、もとの変異が劣性であること、対応する領域のコスミドが存在することが一般的に必要である。コスミドが分離されていないcosmid gap領域でもYACでcoverされていればYACを用いることも可能である。cosmidもYACも分離されていない領域についてはこの方法は使えない。クローニングに要する時間は、もとの変異の表現型の明瞭さと、マッピングの精度に大きく依存する。この方法は相補クローニングとポジショナルクローニングの組合せということができる。
(2)Transposon tagging
MT3126(mut-2)、RW7097(mut-6)などのmutator strain及びBergerac (BO)株において生ずる変異の多くはトランスポゾンTc1の挿入によるものであり、原理的には、これら変異株中のTc1挿入変異遺伝子はTc1をプローブとするハイブリダイゼイションによってクローン化可能である。ただし、mutator株には数百個のTc1が存在し、その中のどれが目的とする遺伝子に挿入したものかを探すことは大変なので、mutator株の個体の中から表現型によって選んだ、目的とする変異株を野生株N2(30個程度しかTc1を持たない)と5回以上back crossして、余分なTc1を減らす。この株及びN2のDNAをEcoRI等で切断し、Tc1をプローブとしてSouthern hybridizationを行い、N2になくてback crossしたmutantにあるいくつかの断片(バンド)を同定する(図3a )。これらのどれかが目的とするTc1挿入遺伝子である。目的とする遺伝子の最も簡単な同定法は、Tc1挿入変異株からTc1の離脱によって再び野生型にもどったrevertantの分離を利用する。この場合、先程のサザン法によって同定したTc1挿入断片のそれぞれを回収し、EcoRVによってTc1を除き、残りのDNAをプローブとして、back crossした変異株、N2及びrevertantのDNAについて再びSouthern hybridizationを行う。N2とrevertantから生ずる断片が同じ大きさで、(back crossした)変異株の断片がこれらより1.6kb(Tc1の大きさ)長ければ、これらが野生型及びTc1挿入を持つ目的とする遺伝子である(図3b)。あるいは、Tc1挿入変異のマップされる位置との対応や、野生型遺伝子断片の変異株への導入によるrescue実験によっても、目的とする遺伝子の同定が可能であろう。revertantを利用して目的とする遺伝子を同定した場合でも、対応する野生型遺伝子の変異株への導入によって、表現型が回復することを確認することが必要である。この方法によるクローニングにも多くの例がある。
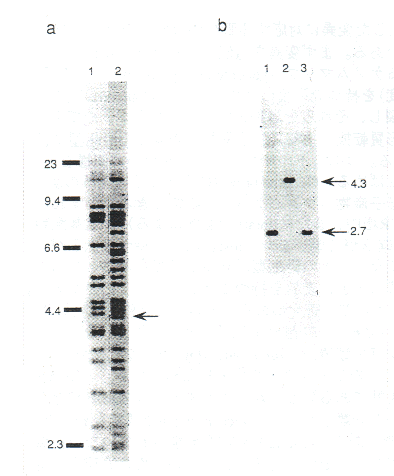
図3.unc-51遺伝子のTc1タギングによるクローン化。(a)N2(lane1)またはback crossしたunc-51変異株FK51(unc-51(ks38::Tc1))(lane 2)の全DNA1mgをEcoR1で切断し、Tc1をプローブとしてSouthern hybridizationを行った。(b)N2(lane1)、FK51(lane2)またはrevertant FK52(lane3)のDNA1mgをEcoR1で切断し、(a)の矢印で示す断片からEcoRVによりTc1を除いたものをプローブとしてSouthern hybridizationを行った。FK51に見られる4.3kbの断片がTc1の挿入したunc-51遺伝子、N2及びreverantに見られる2.7kbの断片が野生型unc-51遺伝子を示す。小倉顕一博士論文(1995)より引用。
(3)PCR(DNA)、RT(逆転写)-PCR(RNA)
ゲノム遺伝子の塩基配列が既知であり、これを基にしてcDNAをクローン化する場合、野生型cDNAの配列が既知であり、変異株のcDNAを調べるためにクローン化する場合、ゲノム遺伝子の一部や上流のプロモーターなどをクローン化する場合などに用いる。塩基配列の決定だけなら、クローン化せずPCR断片をそのまま用いることも可能である。
(4)Hybridization
ゲノム遺伝子をもとにしてcDNAをクローン化する場合、逆の場合などに、cDNAライブラリーやコスミドDNAの断片等をハイブリダイゼイションによって選択し、クローン化する。
(5)ゲノムプロジェクトに基づく遺伝子のクローン化
ゲノムプロジェクトにより、既に多くの推定上の遺伝子の塩基配列が決定されている。コンピューター上のホモロジー検索により、これらの中から自分の興味ある遺伝子を探し出すことができる。これらをクローン化する際には、C. elegansのゲノムDNAやmRNAとPCRまたはRT-PCR法、コスミドの入手、ハイブリダイゼイションなどを用いて行う。
(6)相互作用するタンパク質の遺伝子(cDNA)のクローン化
酵母Saccharomyces cerevisiaeを用いるtwo-hybrid法が現在最も組織的で有効な方法である。文献⑪、⑫はC. elegansにおける利用例である。
文献⑩Durfee et al., Genes Dev. 7, 555-569 (1993)
⑪I. D. Chin-Sang and A. M. Spence, Genes Dev. 10, 2314-2325 (1996)
⑫Ogura et al., Genes Dev., in press (1997)
(7)相補クローニング及び発現クローニング
cosmid rescue法は一種の相補クローニング(complementation cloning)の方法であるが、他生物の突然変異を相補する活性によって、C. elegansの遺伝子(cDNA)をクローン化することも原理的に可能である。実際、酵母S. pombeまたはS. cerevisiaeの変異株を用いてこれが行われている(東大、山本正幸研究室;名大、松本邦弘研究室)。
脊椎動物などでは、多くの遺伝子(cDNA)が、培養細胞等を宿主とする発現クローニング(expression cloning)で得られている。C. elegansについてはまだ殆ど行われていないが、機能が予測されるあるいは検出手段があるものについては可能であり、今後利用の可能性がある。
F.Authodox geneticsとreverse genetics
突然変異(株)の分離から出発し、あるいは既存の変異株を利用して、対応する遺伝子のクローン化などにより、分子レベルの機能の解析をめざす戦略(strategy)を、いわゆるauthodox genetics(正統的遺伝学)を基礎とするやり方と呼ぶことができる。他方、ゲノムプロジェクトから推定された遺伝子、ハイブリダイゼイションやtwo-hybrid法でクローン化された遺伝子など(の配列)から出発し、その生体内での機能を明らかにするためにその遺伝子の突然変異株を分離するあるいは既存の変異株の中から探すというやり方はreverse genetics(逆転遺伝学)と呼ばれている。C. elegansの研究において、現在まではauthodox geneticsに基づく方法が圧倒的に多く用いられ、また成功を収めている。しかし、ゲノムプロジェクトや、今までの研究の進展の結果として、reverse geneticsの必要性、重要性が増しており、両者が共に必要な場合も多くなっている。変異株の表現型の解析、遺伝子発現の解析など両者に共通に必要な点も多い。
(1)Authodox geneticsに基づく研究の方法
1)突然変異株の分離:
解説2(細野)
⑬S. Brenner, The Genetics of Caenorhabditis elegans. Genetics 77, 71-94 (1974)
文献①、Chapter 2(R. K. Herman)
文献②、Chapter 4(Johnsen and Baillie)
文献④、Chapter 2(P. Anderson)
2)マッピングと解析
実習2(細野、森、古賀)
文献⑬
文献④、Chapter 4(B. D. Williams)
3)遺伝子クローニング:前項E参照(主として(1)、(2))。
(2)Reverse geneticsの研究方法
1)当該遺伝子についての突然変異株の分離(遺伝子破壊)
解説3:遺伝子破壊(安達)
G.遺伝子発現の解析
(1)レポーターまたはタグ(tag)との融合タンパクまたは融合遺伝子の発現:実習4(GFP、lac Z、三谷)
(2)抗体染色(In situ immunostaining):実習5(香川)
(3)In situ hybridization of (m)RNA:解説4(小原)
(4)Northern hybridization of RNA
(5)Western blot
(6)Electron microscopic immunocytochemistry(免疫電顕):文献④、Chapter 17, p425-428(D. H. Hall)
C. elegans遺伝子の発現する部位及び時期を調べるには、多くの場合(1)、(2)、(3)のいづれかの方法が用いられている。方法(1)は最も容易で、よく使われる。また、GFP融合タンパクまたは遺伝子の場合、生きた個体で観察可能であって、発現細胞の同定について最も優れている。しかし、人工的融合遺伝子を導入して調べるため、いろいろなartifactが生じ、本来C. elegansがもつ遺伝子(endogenous gene)の発現と異なるパターンとなる可能性があり、注意が必要である。特にプロモーターとの融合遺伝子の発現には問題が多いといわれ、融合タンパク(translational fusion)の発現の方がよい。HA(インフルエンザウィルスへマグルチニン抗原)、Myc、FLAGなどのタグとの融合タンパクも発現の解析によく使われ、細胞内局在部位に関してはGFPやlac Zとの融合タンパクよりもartifactが少ないと考えられる。方法(3)は、広く遺伝子(mRNA)に対して、用いる方法の共通性が高いこと、及び本来の遺伝子の発現を調べる点が優れているが、多くの遺伝子の最終産物であるタンパク質を調べない点に問題がある。(2)の抗体染色は、(1)、(3)の方法の問題点が無く、その意味で最も優れている。しかし、標本を固定するため、GFP融合の場合よりも発現細胞の同定がやや難しい。また、抗体の調製が必要なこと、及び抗体の特異性が問題である。
方法(4)、(5)の主目的はmRNAやタンパク質の大きさや種類の解明であり、C. elegansの場合(1)~(3)と異なり、普通発現部位の解析には使えない。発現時期については解析可能である。
多くのタンパク質について、それを発現する細胞だけでなく、細胞内の存在部位が問題であり、今後ますますその解明が遺伝子機能の解明にとって重要になると思われる(例:⑭Simske et al., Cell 85, 195-204, 1996)。(1)、(2)の方法(特に(2))は、タンパク質の細胞内局在を調べるためにも有用であるが、C. elegansの細胞が比較的小さいこともあり、その局在性の分解能は必ずしも十分ではない。方法(6)を用いることが望ましいが、まだ用いられている例が少ない(例:⑮Ristoratore et al., J. Submicrosc. Cytol. Pathol. 26, 437-443 (1994))。
方法(1)のartifactの1つは、導入遺伝子が、多くのプラスミドDNAがつながってできたextrachromosomal arrayの状態になっていることによる。このarrayは、randomに細胞から失われる可能性がある(→遺伝的モザイク解析)が、その頻度は低く、存在していてもその細胞で発現しない、あるいは発現が弱い場合(mosaic expression)がしばしばある(文献④、Chapter 19, Mello & Fire)。このextrachromosomal arrayが、染色体に組み込まれた系統を選択して用いる(解説7 遺伝子の染色体への挿入(三谷))ことにより、安定になり、遺伝子の発現も強く強くなることがある。しかし、挿入によっても、モザイク的な発現は必ずしも解消されず(文献④、Chapter 19, p478)、その原因はarrayに由来する重複構造(tandem repetition)であり、これは遺伝子導入の際、C. elegansのゲノムDNAなどを過剰に加えることによって防げるといわれる(A. Fire et al., 11th International C. elegans Meeting, Madison, 1997)。
H.遺伝子機能の解析方法
(1)突然変異
機能喪失(loss of function, null)、機能低下(reduced function)、機能増大(gain of function or activated)などの突然変異の表現型によって、当該遺伝子の機能を推定する。機能を詳細に知るためには、いろいろな種類の変異を調べることが必要であり、特に機能を完全に失ったnull変異の表現型が重要とされる。
(2)変異表現型の導入遺伝子発現による回復(rescue)(実習3)
対応する野生型対立遺伝子の導入により、変異株の表現型が回復することは当然であるが、その遺伝子の機能が他生物のクローン化され、分子レベルで機能の分った遺伝子と相同と考えられる時、その導入による表現型の回復を見れば、機能解析に役立つ。例:⑯Vaux et al. Science 258, 1955-1957 (1992)。
(3)遺伝子発現部位及び時期の解析(→前項G)
(4)導入遺伝子の過剰(強制)発現、異所発現
野生型ゲノム遺伝子などをextrachromosomal arrayなどとして導入し、過剰発現させ、その効果を調べる(機能増大突然変異と同様な意味を持つ)。また、細胞特異的プロモーター、熱誘導プロモーターなどとcDNAの融合遺伝子を異所発現させることにより、しばしば当該遺伝子の機能をはっきり示すことができる(例:⑰Hamellin et al., Nature 364, 328-330 (1993))。
(5)遺伝子発現の抑制
1)Dominant negative 変異遺伝子の導入、発現:例=⑱Ogura et al., Genes Dev. 8, 2389-2400 (1994)
2)アンチセンス(anti-sense)RNAの発現:文献⑲A. Fire et al., Development 113, 503-514 (1991)
3)Microinjection of RNA:in vitroで転写したアンチセンスRNAを生殖巣に注入することにより、子孫において当該遺伝子の発現が阻害される例が最近多数報告されている。驚くべきことに、多くの場合この効果は10世代程度までしばしば伝えられ、またセンスRNAでも同様な効果があるという(Driver et al., Abstracts of 11th International C. elegans Meeting, pp118, (1997))。
4)リボザイム等:C. elegansではまだ成功例が無い。
(6)遺伝子産物(タンパク質)のin vitroでの機能解析
大腸菌、哺乳動物・昆虫などの培養細胞などでタンパク質や融合タンパクを発現させ、in vitroで生化学的な解析を行う。抗体の作成のための抗原も多くの場合このような異種発現系を利用して得ている。
(7)遺伝子の培養細胞や異種生物での発現と機能解析
C. elegansの培養細胞系はまだ開発されていないが、哺乳動物や昆虫の培養細胞、アフリカツメガエルの卵母細胞、酵母などでC. elegansの遺伝子を発現させ、その細胞を遺伝子機能のin vivo解析に用いることができる。
例:⑳Brundage et al., Neuron 16, 999-1009 (1996)
21 Komatsu et al., Neuron 17, 707-718 (1996)
酵母(S. cerevisiaeまたはS. pombe)の突然変異を相補(rescue)するC. elegansの遺伝子のクローン化も行われている(E(7)参照)。
(8)相互作用の解析
1)遺伝的相互作用:suppressor mutation、enhancer mutation、synthetic mutationなどの分離と解析。
2)two-hybrid法:遺伝子クローニングだけでなく、相互作用部位の解析等にも利用可能(項目E(6)参照)。
3)in vitro binding、in vivo binding(免疫沈降など)
(9)抗体などによるタンパク質機能の阻害
抗体を生殖巣や細胞に注入やParticle gun等により導入し、その効果を見ることのできる可能性がある。
(10)薬剤の利用
行動、神経機能、dauer幼虫形成などにおける遺伝子機能の解析に、多くの生理活性物質、阻害剤などが用いられている。
I.遺伝子機能部位(細胞)の解析
細胞自律的(cell-autoromous)な遺伝子については、それが機能する細胞は発現する細胞に含まれるはずである。しかし、発現していても機能しているとは限らない。従って、厳密に遺伝子が機能する細胞を同定するには、いわゆる発現の解析とは異なる方法論が必要である。C. elegansにおいては、その一般的方法は遺伝的モザイク解析である。
文献①、Chapter 2, section VI(R. K. Herman)
④、Chapter 6(R. K. Herman)
22 Koga & Ohshima, Development 121, 2655-2666 (1995)
J.細胞機能の解析
(1)Laser ablation:解説6(森)
(2)Toxic geneの発現による細胞の選択的殺傷
ジフテリア毒素(DT-A)などの異種遺伝子、C. elegansのdegenerin遺伝子などを、細胞で発現または過剰発現させることにより、特定の細胞(群)を殺せる可能性がある。この場合、細胞特異的プロモーターを必要とする。
(3)突然変異
細胞系譜、programmed cell deathなどの遺伝子の突然変異により、一群の細胞を生じさせなくし、あるいは殺し、その効果を調べることが可能である。
(4)電気生理学的方法
文献:23 Raizen and Avery, Neruron 12, 483-495 (1994)
24 Li et al., Proc. Natl. Acad. Sci. 94, 5912-5916 (1997)
K.機器
(1)実体顕微鏡
1)Nikon SMZ 1B,透過照明付。実習用(1人1台),定価19.6万円。
2)Leica MZ12 (8×~100×)(研究室備品)+マクロ蛍光装置Leica GFP(実習用デモ)。価格約91万+110万円。
3)Leica MZAPO(8×~80×)+マクロ蛍光装置GFP+モニター(Sony)。3436室,定価約300万円(顕微鏡180万円)。
4)Zeiss Stemi 2000C(6.5×~50×)価格664,000円など(実習用及び3436室)。
5)Leica WILD M8, M5A(6×~50×)(3436室、旧型)。
(2)正立顕微鏡
1)Zeiss Axioplan 2:微分干渉(DIC),蛍光装置,100W蛍光光源,対物レンズ100×or 63×など。2台,実習用デモ機(3435室にも1台)。1台には、CCD Video Camera Module (Sony XC-77CE)+Camera Adapter(Sony CMA-D2)+モニター(Sony PVM-1492Q)がついている(微弱蛍光検出用)。Axioplan 2(微分干渉,蛍光仕様)の標準価格541.7万円。
2)Zeiss Axiophoto:上と同様、本格的写真撮影装置組み込み。1台、実習用デモ機。
3)Nikon Eclipse E800:DIC,蛍光光源及び蛍光用CCDカメラHigh Gain Color Camera HCC-600(フローベル),モニターSony PVM-1455MD,60×(dry)等対物レンズ付。実習用デモ機。価格約500万円?(フル装備顕微鏡定価約360万円)。
4)Nikon Eclipse E600+Color Chilled 3CCD Camera C5810(浜松フォトニクス)+モニター。3435室,価格約500万円。上のデモ機と同様。
5)Zeiss Axioskop (DIC)(研究室備品)+Micro Point Laser Interface System(Photonic Instruments, Inc,標準価格425.7万円)+Zeiss Color Chilled 3CCD ZVS-3C75DEC(199万円)+モニター Sony PVM-1455MD(実習用デモ)。解説6用。
(3)倒立顕微鏡、注入装置
1)Zeiss Axiovert S100。Griding(回転)stage,DIC,Micro Manipulator(Eppendorf5171),Transjector(Eppendorf5246)、CCD Color Video カメラ(Sony DXC-C1, 1/2")+モニター(Victor)付。実習3用デモ機。Axiovert 100,Manipulator,Transjectorの価格計約700万円。
2)Nikon TE300。DIC、Micromanipulator(Narishige MMN-1, MO-202),3CCD Color Video Camera(Victor KYF55B),モニター(Sony, PVM14M4J, 14")付。実習3用デモ機。顕微鏡定価約260万円。
3)Ziss Axiovert 35(旧型)。DIC, Micromanipulator(NarishigeMN-2, MO-202)付。分子遺伝3428室、実習3にも使った。
4)Confocal Microscope, Zeiss LSM410(顕微鏡はAxiovert 135M)3435室(生物学科備品)。定価4,000万円程度以上。
(4)ワークステーション、パソコン(実習6)
1)Sファミリーエンジニアリングワークステーション 富士通S-4/LX,3435室,価格193万円(1993年度)。
2)Personal Computer, Apple Power Mac 7500/100。16MB-RAM/HD1000/CD-ROM M3102J/A,Sony17型ディスプレイCPD-17SF8付。3435室。価格41万円(1995年度)。